带你认识儿童罕见病
发布时间:2023-04-03 15:03来源: 未知梅斯医学与北海康成制药有限公司合作,自2021年起就建立了围绕黏多糖贮积症Ⅱ型疑似患者筛查的医生教育社群,至今已有近4000名儿科医生在此了解了这一儿童罕见疾病。

2023年3月31日晚7点,我们在社群内邀请到了来自安徽医科大学第二附属医院儿童内分泌科主任刘德云教授为我们再次介绍了黏多糖贮积症Ⅱ型的特征及筛查方式。
黏多糖贮积症Ⅱ型(MPSⅡ型)又称为Hunter(亨特)综合征,是由于艾杜糖醛酸-2-硫酸酯酶(iduronate-2-sulfatase, IDS)基因变异引起的IDS活性缺乏,使硫酸皮肤素(dermatan sulphate, DS)和硫酸类肝素(heparan sulphate, HS)不能被完全降解,从而贮积在各种组织、器官的溶酶体中,引起细胞和组织结构、功能改变,进而导致多器官、多系统的结构和功能异常。
黏多糖贮积症Ⅱ型为X连锁隐性遗传疾病,通常男性患病,女性多为携带者。本病患病率存在种族和地区差异,亚洲人群患病率相对高于西方人群,如韩国、日本、中国台湾活产新生儿患病率分别为0.74/10万、0.84/10万、1.07/10万,而多数欧洲国家活产新生儿患病率为0.13/10万~0.71/10万。
黏多糖贮积症Ⅱ型患者出生时多表现正常,随着年龄增长症状逐渐明显,表现为多系统受累,症状进行行加重,可严重致伤、致残。
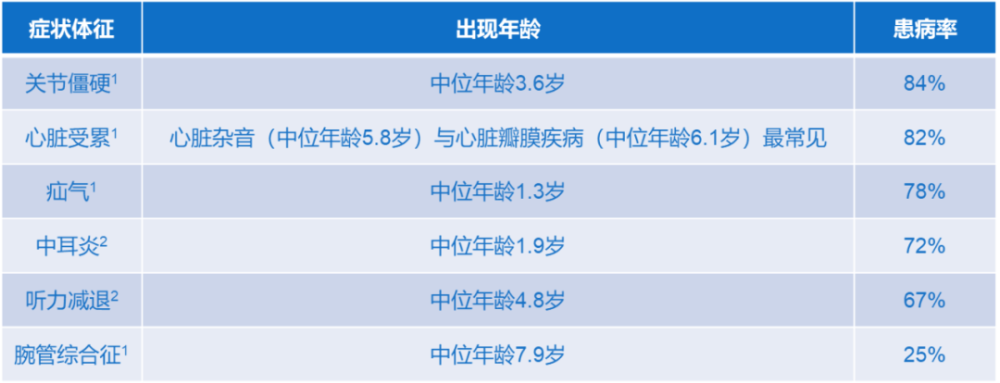
黏多糖贮积症Ⅱ型患者常见的症状和体征包括:
• 面部畸形:面部粗陋、前额突出、眉毛浓密、鼻梁低平、鼻翼肥大、唇厚、舌大、牙龈厚等。
• 腹部症状:疝气,由于肝脏和脾脏增大导致的腹部膨隆。
• 呼吸道症状:反复上呼吸道感染,尤其是耳部感染;睡眠呼吸暂停。
• 骨骼关节障碍:X线检查可见多发性骨发育异常,包括异常的骨增厚及手部关节、肩部关节和肘部关节的不规则骨骺骨化;腕管综合征。
通常由于患儿早期出现的症状不同,而就诊于各个儿童相关科室,调研报告称,通常会经过5年就医,误诊超过7次;儿科医生加强对该疾病的了解,将有效帮助患者减少误诊机率,更早接受合适的治疗。
常就诊科室MPSⅡ高疑似患者的
临床表现及其筛查路径
儿科眼里的MPSⅡ患者
儿科就诊高疑似MPSⅡ患者的临床表现【8】
患儿在儿科就诊的症状较为普遍,没有特异性,需要关注相关的病史、症状或体征,合并发生时需要继续进行筛查。
患儿在儿科就诊时常见的需要关注的病史及合并症状或体征:
•呼吸、耳鼻喉系统:反复上呼吸道感染,听力减退或丧失,腺样体/扁桃体肥大,反复耳部感染;
•关节或骨骼:关节僵硬/爪形手/骨骼畸形/腕管综合征,关节活动受限;
•皮肤和面容:大片蒙古斑,面容粗陋;
•腹部:腹部膨隆,肝和/或脾肿大;
•外科表现:反复多次手术史,尤其是疝修补术,腺样体/扁桃体切除术,鼓膜造孔术;
•神经系统:语言发育迟缓/认知障碍,好动。

